





聚乙烯醇的热降解及热塑加工改性研究进展
江献财 , 董海亚 , 谢静思 , 张 熙* , 代 华
(高分子材料工程国家重点实验室 , 四川大学高分子研究所, 成都 610065)
关键词:聚乙烯醇;热降解;增塑;热塑加工
PVA 是一种性能优异, 用途广泛的水溶性高分子材料。由于其良好的水溶性、成膜性、粘接性 、乳化性和阻隔性能, 因此被广泛应用于纤维 、薄膜、粘接剂、造纸助剂等领域[ 1] 。近年来研究证明[ 2 ~ 4] , PVA 是一种可以完全生物降解的合成高分子材料, 这使其应用范围进一步扩大。但PVA 熔融温度在 220 ~ 240 ℃之间 , 与分解温度接近, 给PVA 的热塑加工成型带来了困难。基于此原因, 常用的 PVA 材料的成型方法均为溶液成型法, 如溶液纺丝、溶液流延成膜等 。溶液加工成型需经历溶解和干燥过程, 存在工艺复杂 、成本高、产量低等缺点。基于溶液加工成型法的 PVA 仅能制备薄膜、纤维等低维制品或用作助 、辅材料。热塑加工相比溶液加工具有工艺简单 、能耗低、效率高 、成本低等优点 。若用挤出、注塑等热塑加工方法则可制备 PVA 三维制品, 从而拓展PVA 的应用领域。因此实现PVA 的热塑加工具有重要的意义。本文对 PVA 的热降解行为和近年来在 PVA 热塑加工改性技术方面的研究进展进行了综述, 希望有助于PVA 的热塑加工新技术的研究。
1 PVA的热降解及稳定
PVA 的热稳定性不高, 其分解温度为 200 ~ 250 ℃。在熔融加工过程中, PV A 受到剪切力和高温两方面的作用, 其中高温是引起PVA 降解进而限制PVA 熔融加工的主要因素。PVA 的降解反应受其结构(如醇解度等)和环境的影响较大 。在不同结构PVA 的热降解研究方面, H olland 等[ 5 , 6] 分别对完全醇解PVA (无醋酸酯基团)和聚醋酸乙烯酯(P VAc)在惰性气氛下的热降解行为进行了研究 。他们通过对 TG-F TI R 联用方法所得的研究结果分析发现, PVA 在第一步脱除羟基生成小分子水后, 主链上并未发现共轭双键的存在, 因此推测羟基脱除反应是在主链上随机发生的。生成的双键会向相邻的羟基转移形成烯醇式结构。烯醇式结构并不稳定, 会发生与酮式结构的互变现象(见图 1a)。PV A 主链的断裂是通过形成一种六元环的过渡状态而随机断裂的, 生成的降解产物主要为乙醛, 也有不饱和的醛、酮类物质(见图 1b , c)。在固体状态下, 由于PVA 链的低活动性, 难于形成六元环的结构, 因此只发生链上羟基的脱除反应。通过对聚醋酸乙烯酯(PV Ac)降解反应研究发现, PVAc 的降解过程中有自动加速现象, 脱除醋酸酯基团生成的双键能促进 PVAc 主链上相邻醋酸酯基团的脱除, 起到加速热降解反应的作用。对于部分醇解的 PVA , 由于主链上含有醋酸酯和羟基两种基团, 其降解行为更为复杂, 热稳定性受醇解度的影响较大。醇解度越低, 残余的醋酸酯基团含量越多, 对 PVA 的热降解促进作用也越大, PVA 的热稳定性则越低。部分醇解的PVA 在第一步脱除反应中会生成醋酸和水两种小分子。主链上残余的羟基会受到醋酸小分子的质子化作用, 形成一种更容易脱除的—OH2 + 结构。
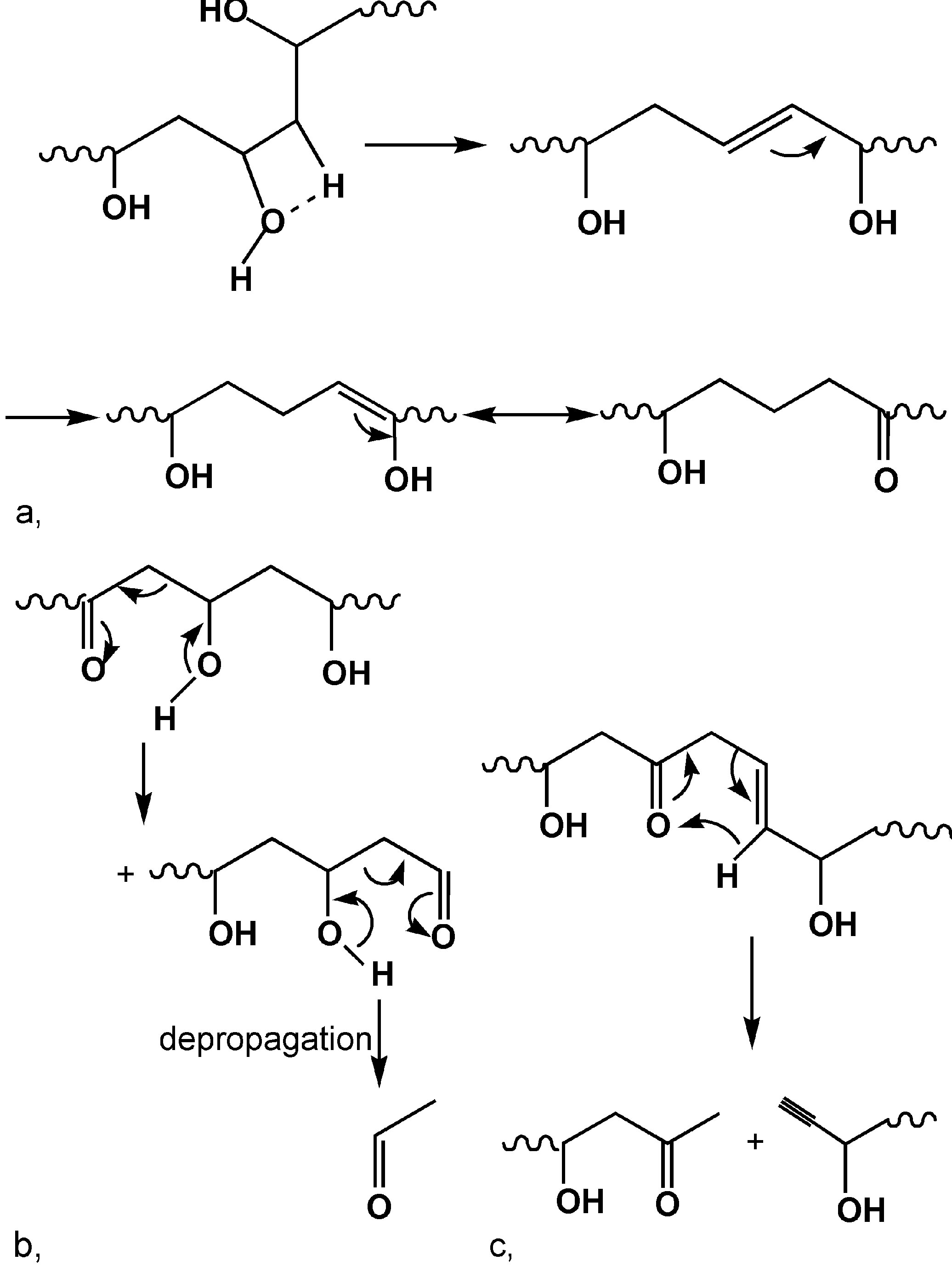
图 1 PV A 在熔融状态下的热降解过程
a.烯醇-酮式结构互变;b.导致链断裂的氢原子转移;c.生成炔端基的氢原子转移[ 5]
PVA 所处环境对其热降解影响显著。一般认为PVA 在惰性气氛下的降解反应遵循脱除机理, 分为两步进行[ 7] :首先是水和醋酸等小分子的脱除反应 ;其次是 PVA 主链的断裂生成醛、酮、呋喃 、苯及苯的衍生物等。在空气中 PVA 的降解也基本遵循以上的两步脱除机理, 但在两步反应中均有氧化反应的参与, 机理更为复杂。 Tho ms 等[ 8] 采用 TG-F TIR 联用的方法初步研究了在空气中和无氧密闭条件下PVA 的降解机理。TG 曲线显示PVA 在空气气氛下的前期失重速率比在密闭无氧条件下有明显下降, 充分显示了在第一步脱除反应中有氧的参与。由于有氧气的参与, 在空气气氛下第二步的断链热解反应速率则明显增加。
采用一些预处理方法和外加助剂能提高PVA 的热稳定性。无机物的加入能明显地提高 PVA 的热稳定性, 使得PVA 热降解中的第一步小分子脱除反应在更高的温度下发生, 从而使得在羟基和醋酸酯基团脱除的同时就有主链断裂反应的发生[ 9] 。
加入增塑剂和润滑剂能降低PVA 的熔融温度和熔体粘度, 这有助于提高PV A 在熔融加工中的稳定性, 但并不能完全克服PVA 在熔融加工中的热降解问题。Alexy 等[ 10 , 11] 研究了PVA 熔融加工热稳定性对PVA 预处理时溶液pH 值的依赖性, 结果表明对于PV A 热塑加工最为稳定的预处理溶液 pH 值在4 左右。
从PVA 的降解机理可以看出, PVA 的热降解会生成醋酸小分子, 醋酸能质子化PVA 主链上的羟基![]() 基团, 形成更容易脱除的—OH + 结构, 加快PVA 的热降解。加入碱性物质能除去 PVA 降解生成的醋酸分子, 因此加入碱性物质能提高PVA 的热稳定性。在此思路上Alexy 等将碱性无机盐类物质加入到PVA 中提高其热稳定性, 结果证明加入无机碱能提高 PVA 的热稳定性, 其中M g(O H )2 能最大程度的推迟PVA 的降解, CaO 的加入能最大程度地抑制 PVA 的降解。在 PVA 混合体系中经常使用的硅土, 由于其酸性性质, 则能明显地降低PVA 热稳定性。
基团, 形成更容易脱除的—OH + 结构, 加快PVA 的热降解。加入碱性物质能除去 PVA 降解生成的醋酸分子, 因此加入碱性物质能提高PVA 的热稳定性。在此思路上Alexy 等将碱性无机盐类物质加入到PVA 中提高其热稳定性, 结果证明加入无机碱能提高 PVA 的热稳定性, 其中M g(O H )2 能最大程度的推迟PVA 的降解, CaO 的加入能最大程度地抑制 PVA 的降解。在 PVA 混合体系中经常使用的硅土, 由于其酸性性质, 则能明显地降低PVA 热稳定性。
2 聚乙烯醇的热塑加工改性技术
目前涉及的实现PVA 的热塑加工的方法可概括为如下五种:直接加入增塑剂 、共聚改性、控制 PVA醇解度和聚合度 、后反应改性 、与其它高分子材料复配改性 。
2.1 应用增塑剂的聚乙烯醇热塑加工技术
加入小分子物质或低聚物增塑剂是实现PV A 热塑加工最为常用的一种方法。加入的小分子物质或低聚物可与 PVA 分子链上的羟基形成氢键, 从而减少PVA 相互之间形成氢键的概率, 同时小分子物质还可以起到润滑剂的功效, 上述作用可降低PV A 的熔点, 改善 PVA 熔体流动性, 从而使 PVA 可在较低的温度下具有较好的流动性、实现热塑加工。从目前的研究情况看, 加入小分子或低聚物是实现PVA 热塑加工的最为有效和直接的方法。PVA 热塑加工的增塑剂主要是含有多羟基的小分子物质或者含有能与PVA 形成氢键复合的醇胺类物质、酰胺类物质。国内外文献报道的塑化改性剂有:水及甘油等小分子多元醇类物质、己内酰胺 、醇胺类物质及分子量较低的聚乙二醇(PEG)低聚物。
最为常用的多元醇类增塑剂是高沸点的甘油。Jang 等[ 12] 研究了甘油对聚乙烯醇的熔融和结晶行为的影响, 研究表明:甘油能增加聚乙烯醇的链段活动性 、减小结晶区域 , 从而降低熔点。但甘油的增塑效果随着甘油含量的增加而逐渐减小, 直至发生相分离时增塑效果急剧减小。对完全醇解的 PVA 相分离发生的甘油加量为 40phr(份 , 每一百份基体中添加的增塑剂份数), 而不完全醇解的PV A 相分离发生在 65phr 。单独使用甘油难于实现PVA 的热塑加工, 且存在甘油极易从PVA 基体中析出, 使制品发脆的问题。
水也可作为PVA 的增塑剂使用, 但其沸点低, 加工中易蒸发使制品含有气泡。在水复合增塑体系的研究方面, 王琪教授[ 13 ~ 16] 以水为主增塑剂, 通过选用与PVA 有互补结构的己内酰胺与水组成复配改性剂, 破坏了PVA 自身分子内和分子间氢键, 抑制了PVA 的结晶, 降低了熔点, 并通过 PVA 与己内酰胺和水分子间的氢键, 改变了水在PVA 中的存在状态, 减少自由水含量, 增加了可冻结合水和非冻结合水含量, 实现了水在PVA 中的过热化, 在加工温度下不产生气泡, 在通用熔融挤出设备上实现了 PVA 1799 的热塑加工, 进而实现了其吹塑成膜和熔融纺丝。采用适当的后处理可制得性能优良的吹塑薄膜和熔纺纤维[ 17 , 18] 。其工艺为先将能与 PVA 形成互补结构的己内酰胺与水复配混匀后加入到经计量的 PVA 中混匀 、溶胀 , 得到可熔融挤出的热塑性PVA , 再熔融纺丝或吹塑成膜。[ 19]
Anthony 等 发明了一种通过热塑加工制备PVA 薄膜的方法。其主要原料PVA 微粉的醇解度为74~94, 质量分数为 80~90, 而增塑剂的质量分数为10~20。增塑剂中有一部分是水, 另外一部分可能是一种或几种多元醇, 如甘油、乙二醇、二乙二醇、三乙二醇、分子量为 200 以下的 PEG 等。结果发现共混改性后的PVA 可以通过挤出机进行吹塑, 其加工温度范围是 185 ~ 210 ℃。
在醇胺类复合增塑改性剂的研究方面, 项爱民等[ 20] 筛选出具有较好增塑效果的醇胺类复配改性剂,降低了PVA 的熔融温度和熔融焓, 改善了流动性, 实现了 PVA 1788 的干法造粒和吹塑成膜。FTIR 研究表明醇胺类改性剂与PVA 分子间发生了强烈的相互作用, 以更强的分子间键合取代了 PVA 本身的键合作用。成膜助剂的加入能明显改善PVA 的加工流动性。
在醇基复合增塑改性剂的研究方面, 王婧 、苑会林等[ 21 , 22] 将多元醇低聚物和低分子醇复配增塑改性PVA 1788 制备了PVA 薄膜。其工艺为将 PVA 、润滑剂 、开口剂按一定比例加入温控高速混合机内, 80 ℃搅拌 0 .5h 后分别用单螺杆挤出机和双螺杆挤出机挤出造粒, 然后进行吹塑成膜。研究发现单独两种增塑剂均无法实现 PVA 的热塑加工。而当两种增塑剂复配后, 其增塑效果大大增加, 可明显改善PVA 的加工流动性, 当复合增塑剂用量在 25phr 以上时, PVA 可被较好的增塑, 熔融温度趋于定值。不同醇解度的 PVA 树脂在改性后均能熔融挤出加工吹塑成膜。
在增塑剂-稳定剂复合改性的研究方面 , 李亚东等[ 23 , 24] 以 PEG 为增塑剂, Mg(O H )2 为稳定剂, 通过单螺杆挤出造粒后注塑成型制备了PVA/ PEG/ Mg(O H)2 复合材料。其结果研究表明 :当 PEG 用量为 20phr 时, 体系的熔融温度降低了接近 10 ℃左右 , 可以有效降低复合体系的熔融温度。Mg(O H)2 可增加 PVA 和PEG 之间的界面结合力, 且可吸收PVA 熔融加工中由于水分的存在而发生水解反应脱下的小分子醋酸, 起到提高复合体系的热稳定性和熔融塑化效果。
从目前的研究来看, 加入小分子或低聚物是实现PVA 热塑加工的最为有效和直接的方法, 国内外对增塑剂的报道也较多。PVA 热塑加工的增塑剂主要是含有多羟基的小分子物质或者含有能与PVA 形成氢键复合的醇胺类物质。从报道的效果来看, 单一增塑剂的加入均难以实现PV A 的热塑加工, 将不同增塑剂复合后加入PVA 中增塑效果更好。
2.2 共聚改性技术
近年来, 对共聚改性PVA 的研究开发也日益增多。共聚改性技术是通过选择可共聚单体与醋酸乙烯进行溶液共聚, 制得醋酸乙烯酯共聚物后在甲醇中醇解、干燥从而获得可热塑加工的共聚改性PVA[ 25] 。该技术主要是通过在PVA 主链或侧基上引入作用力较弱的单体结构单元, 减弱其分子间和分子内作用力, 达到降低熔点的效果。乙烯基单体和醋酸乙烯的共聚反应活性相近, 最有可能生成典型的无规共聚体[ 26] , 是 PVA 共聚改性的首选单体。以乙烯基单体改性PVA 可有效降低熔点。日本三菱化学、合成化学、佳友化学、电气化学等公司均有此方面的专利[ 27] 。日本可乐丽公司合成了可热塑加工的PVA , 该PVA 含有碳原子数小于 4 的 α-烯烃单元及/ 或乙烯醚单元, 熔点为 160 ~ 230 ℃。日本合成化学公司开发的 AXPVA 共聚物薄膜, 其成型加工性能优异, 熔点在 200 ~ 210 ℃, 成型加工温度在 210 ~ 230 ℃, 其热塑加工性能也是通过与其他单体共聚改性而获得[ 28] 。
目前使用的最成功的一类共聚改性物当属乙烯/ 乙烯醇共聚物(EV OH )[ 29] 。EVOH 是由乙烯-醋酸乙烯共聚物经皂化或部分皂化反应而得的醇解产物, 是聚乙烯醇的一种改性产物, 生产工艺流程与 PVA 相似。EVOH 是一种集乙烯聚合物的加工性和乙烯醇聚合物的气体阻隔性于一体的新型高分子合成材料。EVO H 的显著特点是对气体具有极好的阻隔性和极好加工性, 另外, 透明性、光泽性、机械强度、伸缩性 、耐磨性、耐寒性和表面强度都非常优异 。目前 EVO H 已经成为应用最多的一类高阻隔性材料, 与聚偏二氯乙烯(PV DC)和聚酰胺(PA)并称为三大阻隔材料 。
2.3 PVA 后反应改性技术
PVA 后反应改性技术是通过PV A 分子链上含有的高活性仲羟基进行化学改性, 在PV A 分子链上引入其它可降低 PVA 的规整度和提高热稳定性的结构单元, 从而改善PVA 的热塑加工性能, 实现热塑加工。
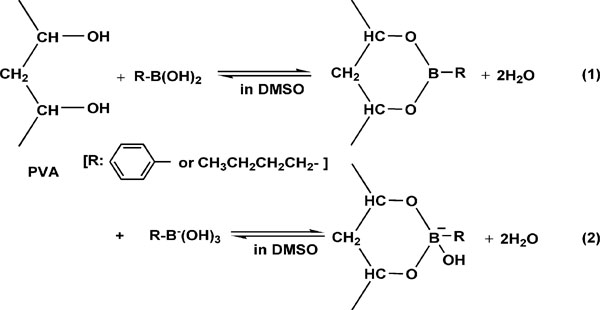
高峻等[ 30] 通过硬酯酸与PVA 的酯化反应, 使 PVA 分子中部分羟基酯化, 引入硬脂酸基侧链。酯化后的PVA 结晶度和熔融温度降低, 热稳定性和耐水性明显提高。Nishimura 等[ 31] 用正丁基硼酸和苯基硼酸与PVA 形成络合物把PVA 转化为熔融流动性的衍生物, 然后将衍生物进行纺丝并用热水处理除去硼酸络合物得到PVA 纤维, 从而实现PVA 熔融纺丝。研究表明, 烷基硼酸络合物能有效地降低PVA 的熔融温度和熔融焓, 提高分解温度。相关反应如上所示。
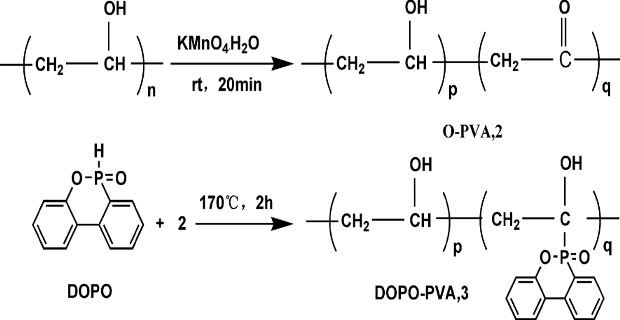
Liu 等[ 32] 采用KMnO4 氧化法使PVA 主链上含有乙烯酮结构单元, 然后将DOPO 作为亲核试剂与PVA 链上的羰基反应, 将含磷的 DOPO 基团连接到 PVA 主链的碳原子上, 得到的含 DOPO 基团的PVA 具有更好的热稳定性、油溶性和阻燃性能。相关反应如下所示。[ 33]
Harala 等 将不同链长的脂肪酸在熔融状态下与PVA 发生酯化反应以改善PVA 的热稳定性和熔融流动性能。长链脂肪酸可以起到润滑剂的作用并延缓 PVA 的热降解, 从而使得熔融反应得以进行。他们的研究结果表明 :酯化反应程度与设备有关, 其原因可能为反应设备几何形状不同引起的剪切和混合条件的改变, 在双螺杆挤出机中, 己酸和辛酸酯化反应率最高, 因此对降低熔点的效应最为明显, 酯化改性后的PV A 的热稳定性均比纯PVA 要高。
2.4 通过控制PVA 醇解度和聚合度实现热塑加工
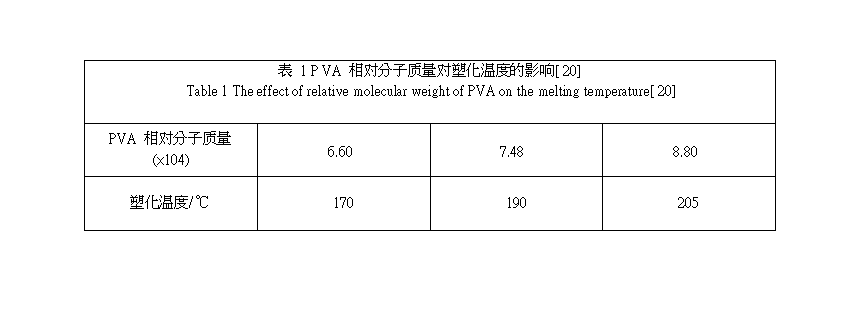
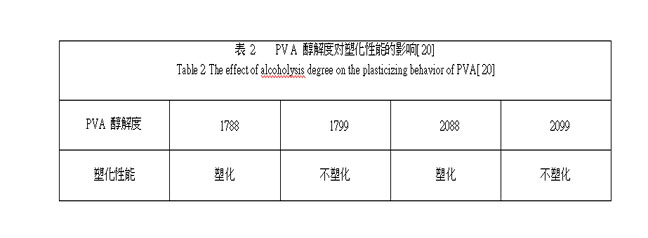
PVA 的性能主要由聚合度和醇解度控制, 因此通过控制聚合度和醇解度可制得不同性能的 PVA 材料, 实现PVA 热塑加工。项爱民研究发现[ 20] , 在相同塑化改性剂用量下, 随着 PVA 相对分子质量的增加, 其塑化温度升高(见表 1);随着醇解度增加, PVA 塑化性能下降(见表 2)。通过制备低聚合度或低水解度的PVA 可实现 PVA 的热塑加工, 这也是目前工业上主要采用的方法。例如, 美国 K URARA Y POVAL CP 系列PVA 树脂的热塑性及水溶性是通过控制PVA 的聚合度和醇解度而获得的, 具有优良的热塑加工性能[ 34] , 可在 170 ℃左右熔融加工 。美国Air Product &Chemical 公司也开发出了以聚合度较低的PVA 为基础的树脂, 该树脂同时具有水溶性、热塑性、生物降解性 , 它可通过挤塑 、共挤塑 、纺丝成形, 可制得适用于食品包装的薄膜 、农用薄膜 、容器及一次性消费用品等。虽然降低 PVA 的聚合度和醇解度是实现 PVA 热塑加工的有效途径, 但降低聚合度和醇解度必然会导致PV A 综合性能的损失。
2.5 与其它高分子材料共混
PVA 与天然高分子材料共混后热塑性能得到改善, 加入小分子增塑剂后使得熔融加工成为可能。天然高分子材料之所以能与PVA 共混提高热塑性能, 是因为它们都带有能与羟基生成氢键的基团, 从而破坏PVA 分子间的强作用力, 起到改性作用。PVA 和天然高分子材料都具有生物降解性能, 因此将PVA 和天然高分子材料共混可制得完全生物降解材料。正是由于以上原因, 采用PVA 与天然高分子材料共混的研究得到了人们的重视。
在有关淀粉 PVA 共混研究方面, 王会才等[ 35] 将淀粉、PV A 及增塑剂按一定比率混合后由双螺杆挤出机造粒后用单螺杆挤出机挤出成片材, 研究了PVA 与淀粉共混体系在不同增塑剂增塑下的共混挤出工艺。研究表明 PVA 形状和醇解度对热塑加工性能均有影响, 片状PVA 1788/ 淀粉用水和甘油溶胀后可以进行挤出, 而与甘油干混后无法进行挤出, 粒状PVA 1788/ 淀粉用水和甘油溶胀后及与甘油干混后均可进行挤出, PVA 1799/淀粉体系无论采用以上哪种工艺均无法进行挤出。增塑剂甘油用量为 40 phr 时对共混体系起到较好的增塑作用, 此用量下体系的拉伸强度和断裂伸长率最佳。目前最成功的 PVA/ 淀粉复合材料是意大利Montedison 集团Novamont 公司开发生产的“ Mater-Bi”品牌。它是由变性淀粉与改性PVA 共混构成的互穿网络结构高分子塑料合金, 与其它塑料合金一样可完全发挥各组分所长, 具有良好的成型加工性 、二次加工性 、力学性能和优良的生物降解性能。目前 , 该公司现已开发出挤出成型用片、吹塑薄膜 、流延薄膜 、注塑制品 、中空容器、玩具等产品 。目前限制淀粉/ PV A 塑料使用的主要问题是淀粉/ PVA 复合材料的亲水性太强, 制品使用过程中性能发生变化且价格偏高。降低成本及改善PVA/ 淀粉材料的疏水性是当前PVA/ 淀粉塑料的研究热点。
蛋白质由于良好的溶液成膜性能和在熔融和溶液状态均具有的良好的流动性而得到广泛的应 用[ 36 ~ 38] 。在蛋白质与PVA 共混研究方面, A lexy[ 39 ~ 42] 等选用胶原水解物与PVA 、甘油共混熔融挤出, 制备出了可生物降解薄膜, 该膜显示出良好的机械力学性能, 且胶原质水解物的加入能促进PVA 膜的生物降解。但相对淀粉和纤维素而言, 蛋白质成本较高, 限制了蛋白质的使用。
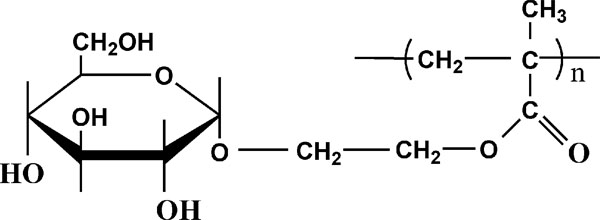
图 2 P oly(G EM A)的化学结构式[ 43]
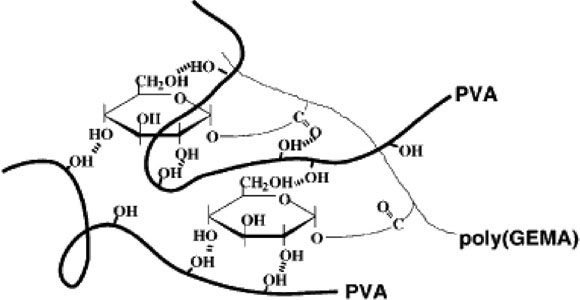
图 3 P VA 与 Po ly(GEM A)的相互作用示意图[ 43]
另外也有将 PVA 和其它聚合物进行共混以改善热稳定的研究的报道。Nishino[ 43] 将糖类衍生物Poly(G EM A)(化学结构见图 2)与 PVA 溶液共混后干燥得到混合物。研究结果表明加入少量的Poly (G EM A), 混合物的分解温度急剧上升, 当 Poly(G EMA )的质量分数增加到 25 ![]() 时, 混合物的分解温度达到 326 ℃, 与PVA 分解温度的差距达到 100 ℃, 加工窗口大大拓宽。混合物的分解温度升高是因为Poly(G EM A)侧链上的葡萄糖基元能与 PVA 分子链上的羟基形成更强的氢键作用, 形成分子复合物(见图 3), 从而抑制了PVA 在高温下的降解。
时, 混合物的分解温度达到 326 ℃, 与PVA 分解温度的差距达到 100 ℃, 加工窗口大大拓宽。混合物的分解温度升高是因为Poly(G EM A)侧链上的葡萄糖基元能与 PVA 分子链上的羟基形成更强的氢键作用, 形成分子复合物(见图 3), 从而抑制了PVA 在高温下的降解。
3 结束语
21 世纪新材料发展非常迅速, 优胜劣汰的竞争将更为激烈。我国是PVA 生产第一大国, 2010 年, 我国PVA 产能将达到 90 万吨, 预计表观消费量 70 万吨。但我国对PVA 的热塑加工的研究并不充分, 在实现PVA 热塑加工的途径上略显单一, 同国外, 尤其是日本和美国, 还是有一段很大的差距, 这大大限制了我国PVA 工业在国际上的竞争力。只有通过加强研究开发工作, 拓宽实现PVA 热塑加工的途径, 突破PVA 因传统溶液加工成型仅能制造纤维 、薄膜或用于助、辅材料的局限, 研制高性能PVA 基材料, 拓宽PVA 材料的应用领域, 才能为我国 PVA 行业注入新的活力, 实现我国由PVA 生产大国向生产强国的转变。
参考文献
[ 1 ] 严瑞瑄.水溶性高分子.北京:化学工业出版社, 1998 , 42 .
[ 2 ] Chiellini E , C orti A , Salvatore D, Solaro R .Prog Polym Sci, 2003 , 28 :963 ~ 1014 .
[ 3 ] M atsumu ra S , Tomizaw a N , T oki A , Nishikaw a K , Toshima K .M acromolecules, 1999 , 32 :7753 ~ 7761 . [ 4 ] Corti A , Solaro R, Chiellini E .Pol ym Deg rad Stab , 2002 , 75 :447 ~ 458 .
[ 5 ] H olland B J , Hay J N .Polymer 2001 , 42 :6775 ~ 6783 .
[ 6 ] H olland B J , Hay J N .Polymer 2002 , 43 :2207 ~ 2211 .
[ 7 ] Gilman J W , Kashiw agi T , van der Hart D L, Proceeding s of th e ACS Division of Polymeric M aterials , Science an d E ngineering , Fall M eeting , 1994 , 71 :28 .
[ 8 ] Thomas P S , Guerbois J P , Russell G F , Briscoe B J .Journal of Th ermal Analysis and C alo rim etry 2001, 64 :501 ~ 508 . [ 9 ] Zheng P , Kong L X .Polym Degrad S tab , 2007 , 92 :1061 ~ 1071 .
[10] Alexy P , Kach ova D, Krsiak M , Bakos D, Sim kova B .Polym Degrad S tab , 2002 , 78 :413 ~ 421 .
[11] Alexy P , Lacik I , Simkova B , Bak os D , Pronayova N , Lip taj T , H an zelova S , Varosova M .Polym Degrad S tab 2004 , 85 :823 ~ 830 .
[12] Jang J , Lee D K .Polym er , 2003 , 44 :8139 ~ 8146 .
[13] 王茹, 王琪, 李莉, 华正坤.高分子科学与工程, 2001 , 17(6):111 ~ 113 .
[14] 王茹, 王琪, 李莉, 敖宁建, 华正坤.塑料工业, 2002 , 30(1):32 ~ 34
[15] W an g R, Wang Q , Li L .Polym In t , 2003 , 52 :1820 ~ 1826 .
[16] Ch en N , Li L , Wang Q .Plast Rubber Compos 2007 , 36 :284 ~ 290 .
[17] Li L , W ang Q , Wang R .J Appl Polym S ci , 2005 , 98 :774 ~ 779 .
[18] Zhang H , W ang Q , Li L .Polym Int , 2009 , 58 , 97 ~ 104.
[19] An th ony T P , M ark G K , Vivian S T .中国, GB :W O2004046229 , 2004
[20] 项爱民, 刘万蝉, 赵启辉, 康智能.中国塑料, 2003 , 17(2):60 ~ 62 .
[21] 苑会林, 马沛岚, 李军.塑料工业, 2003 , 31(9):23 ~ 25 .
[22] 王婧, 苑会林, 马沛岚, 李军.塑料工业, 2004 , 32(7):30 ~ 35 .
[23] 李亚东, 胡卉, 魏松, 曹少魁.郑州轻工业学院学报(自然科学版), 2009 , 24(1):34 ~ 37 .
[24] 李亚东, 白宝丰, 魏松, 曹少魁, 宋华栋.塑料工业, 2008 , 36(12):38 ~ 41 .
[25] 汪宝林.化学工程师, 2006 , 126(3):41 ~ 44 .
[26] Barlow A, et al .J Appl Poly m S ci , 1967 , 11 :2001 ~ 2005 .
[27] 侯双燕, 高绪珊, 童俨.合成纤维, 2009 , 5 :1 ~ 3.
[28] 刘峰, 张康助, 王晓洁.化学与黏合, 2006 , 28(4):253 ~ 256 .
[29] 樊岩, 胡绍华, 章悦庭.化工新型材料, 2000 , 28(2):23 ~ 26 .
[30] 高峻, 雷景新, 李训刚.高分子学报, 2001 , 30(1):118 ~ 120 .
[31] Nishimu ra H , Donkai N , M iy amoto T .J Poly m S ci A :Polym Ch em , 1998 , 36 , 3045 ~ 3050 .
[32] Liu Y L , Chiu Y C .J Polym S ci A :Poly m C hem , 2003 , 41 :1107 ~ 1113 .
[33] Haralabakopoulos A A , T siourvas D , Paleos C M .J Appl Polym Sci, 1998 , 69(9):1885 ~ 1890 .
[34] Yamaura K , Kumaku ra R .J Appl Polym S ci , 2000 , 77 (13):2872 ~ 2876 .
[35] 王会才, 崔永岩.工程塑料应用, 2004 , 32(2):27 ~ 29 .
[36] Vanin F M , S obral P J A , M en egalli F C , Carvalho R A , Habitante A M Q B .Food Hydrocolloids, 2005 , 19 :899 ~ 907 .
[37] S udhamani S R, Prasad M S , Sankar K U .Food Hy drocolloids, 2003 , 17 :245 ~ 250 .
[38] Dicharry R M , Ye P , Saha G , Waxman E , Asandei A D, Parnas R S .Biomacromolecul es 2006 , 7 :2837 ~ 2844 .
[39] Alexy P , Bakos D , Hanzelova S , Kuk olikova L , Kupec J , Charvatova K , Chiellini E , Cinelli P .Polymer Testing , 2003 , 22 :801 ~ 809 .
[40] Alexy P , Bakos D , C rkonova G , Kramarova Z , Hoffm an n J , Julin ova M , C hiellini E , Cinelli P .Polymer Testing , 2003 , 22 :811 ~ 818 . [41] Alexy P , Bakos D , C rkonova G , Kolomaz nik K , Krsiak M .M acrom ol Sym p 2001 , 170 :41 ~ 49 .
[42] C rkonova G, Alexy P , Bak os D , Kolomaz nik K , S imkova B , Precnerova L .M acrom ol Sym p , 2001 , 170 :51 ~ 59 . [43] Nishino T , Kani S , Gotoh K , Nakam ae K .Polymer 2002 , 43 , 2869 ~ 2873 .

