





Rapid synthesis of high strength cellulose–poly(vinyl alcohol) (PVA) biocompatible composite films via microwave crosslinking
1Department of Materials Science and Engineering, Indian Institute of Technology Kanpur, Kanpur 208016, India 2Department of Biological Sciences and Bioengineering, Indian Institute of Technology Kanpur, Kanpur 208016, India 3Centre for Environmental Scie
Amit Kumar Sonker, Kalpana Rathore, Arun Kumar Teotia, Ashok Kumar, Vivek Verma
1 ABSTRACT
The present study describes microwave (MW)-assisted rapid synthesis of biocompatible poly(vinyl alcohol) (PVA) compos- ite films that demonstrate synergy between reinforcement and crosslinking. Bacterial cellulose (5% w/w) nanowhiskers (reinforcement) and tartaric acid 35% (w/w) (crosslinker) are incorporated in PVA to prepare crosslinked cellulose–PVA composite films. The properties of thus prepared crosslinked cellulose–PVA composite films are compared with samples crosslinked with conventional hot air oven heating (CH). Crosslinking by both of the methods reduces water absorption of PVA by around an order of magnitude and improves its thermal stability. An increase in strength from 42 (PVA) to 172 MPa and 159 MPa for MW and CH crosslinked samples, respec- tively is also observed. Although composites prepared using MW and CH show similar properties, MW takes only 14 min compared to 2 h in case of CH. Notably, the prepared composites demonstrate hemocompatibility and cytocompatibility, and may also be explored for biomedical applications.
2 INTRODUCTION
Poly(vinyl alcohol) (PVA)–cellulose composite are becoming the preferred choice as biodegradable plastics,1,2 absorbents,3,4 and scaf- folds for tissue engineering.5–9 Although both PVA and cellulose dif- fer in their origin and properties, both are biodegradable and biocompatible. PVA is a synthetic, water-soluble, thermoplastic polymer obtained by hydrolysis of poly(vinyl acetate).10 Due to its excellent chemical resistance and film-forming properties, PVA is used as support material for three-dimensional printing,11 electro- spinning, and tissue engineering.12–14 Cellulose, on the other hand, is a plant or bacteria derived natural exopolysaccharide with d-glucopyranosyl as basic units. The shape of cellulose fibrils depends on the local environment in which the cellulose is pre- pared.15,16 Although bacterial and plant-derived cellulose is structur- ally similar, bacterial cellulose has higher crystallinity and purity as it is devoid of any lignin or hemicellulose. In addition to that, reduced fiber diameter and open network architecture of bacterial cellulose results in the higher surface area in comparison to that of plant cellu- lose.16,17 Due to these unique properties and biocompatible nature, bacterial cellulose holds its applications in tissue engineering18–20 and as reinforcement material.21–24 The nanowhiskers and nanocrystals synthesized from bacterial cellulose are even more suited as reinforcement due to their high aspect ratio, and improved crystallinity and mechanical properties compared to that of as derived natu- ral cellulose.16,25,26
PVA and cellulose composites are used for various applications in the light of properties aforementioned. Tummala et al. prepared hyperelastic contact lenses using PVA–cellulose nanocrystal hydro- gels.8,9 Anisotropic bacterial cellulose–PVA nanocomposite is pre- pared for biomedical applications.6 PVA–cellulose hydrogel composite is developed for adsorption of heavy metal ions.4 PVA–cellulose nanofibrils superabsorbent are prepared for various types of oils and organic solvents.3
A large number of hydroxyl groups on cellulose and PVA allow good hydrogen bonding that leads to higher strength of the com- posite. Crosslinking PVA and cellulose not only improves mechan- ical strength but also impart better thermal and water resistance to the composite due to the covalent bond formation. Crosslinking hydroxyl groups between PVA chains using dicarboxylic acids,27–30 tricarboxylic acids,31,32 diisocyanates,33 and organosi- lanes34 is demonstrated in earlier studies. As both the PVA and cellulose have similar hydroxyls groups, it is expected that PVA and cellulose can be crosslinked using di–tri carboxylic acid and dialdehydes. For example, Qiu and Netravali showed improvement in thermal and mechanical properties of cellulose–PVA composite by crosslinking them with glyoxal1 and glutaraldehyde.2
Crosslinking is usually performed by conventionally heating (CH) solid physical mixture of cellulose, PVA, and crosslinker in a hot air oven for 2–4 h.28,31 Microwave (MW) irradiation is reported to result in similar mechanical, thermal, and water resis- tance properties of PVA crosslinked with tartaric acid (TA) while consuming one-eighth the period required in CH.35
In the present study, MW is used to develop PVA biocomposite based on the synergistic effect of bacterial cellulose nanowhiskers (BCNW) reinforcement and a TA crosslinking. To show synergy between nanoreinforcement and crosslinking, 5% BCNW concen- tration for nanoreinforcement is taken as described elsewhere.28 Similarly, 35% w/w concentration of TA is used as crosslinker.35 It is expected that the interfacial bonding between BCNW and PVA can be improved by crosslinking BCNW and PVA using TA. In detail, wettability, hemocompatibility and in vitro cell compatibility of the developed biocomposite films are also dis- cussed to show the applicability of this biocomposite in tissue engineering.
3 EXPERIMENTAL
3.1 Materials
PVA [degree of polymerization (1700–1800) and degree of hydrolysis (98–99%)], TA (99%), sodium hydroxide (98%), magnesium sulfate (99%), yeast extract (60%), and bacterial peptone were purchased from Loba Chemie (Mumbai, India). Corn steep liquor was procured from Sigma Aldrich (St. Louis, MO). Polystyrene Petri dishes were purchased from Tarsons Product Pvt. Ltd. (Kolkata, India). Sodium chloride, potassium chloride, disodium hydrogen orthophosphate, and potassium dihydrogen orthophosphate were purchased from Thermo Fisher Scientific (Mumbai, India). 3-(4,5-Dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco’s Modified Eagle’s medium (DMEM)-high glucose, trypsin–ethylene diamine tetraacetic acid (EDTA), and antibiotic (penicillin and streptomycin) solution were acquired from Sigma Aldrich (St. Louis, MO). Triton-X and methanol were procured from Merck (Darmstadt, Germany). Disodium hydrogen phosphate and ammonium sulfate were pro- cured from Fisher Scientific (Hampton, USA). Glucose was pur- chased from Qualigens (Mumbai, India).
Preparation of BCNW. BCNW were prepared by acid hydrolysis of bacterial cellulose pellicles grown using Acetobacter xylinum (ATCC 700178) as described by Sonker et al.28 Bacterial cellulose pellicles were first homogenized using a tissue homogenizer (IKA T 18 digital ULTRA-TURRAX) for 2 h. The homogenized bacte- rial cellulose was acid hydrolyzed by adding 63.5 wt % sulfuric acid with continuous stirring at 45 for 4 h. The acid was neu- tralized with 1 M sodium hydroxide to stop the hydrolysis and extra acid and salts were removed with deionized (DI) water using multiple centrifugation cycles (19 000g for 30 min). The centrifugation cycle was stopped when the supernatant solution turned murky and remained suspended. The bacterial CNW sus- pension was then sonicated for 1 h and stored at 4℃ for further use.
Preparation of Tartaric Acid Crosslinked Films. Physical mix- ture (solid film) of TA–PVA film was prepared as per Sonker et al.35 In brief, 2.5 g PVA was dissolved in 100 mL hot water at 90℃ followed by addition of 875 mg (35% w/w) TA. Crosslinking of solid dried TA–PVA films was performed using either CH or MW irradiation. Crosslinking by CH was performed by heating physical mixture of 35% w/w TA–PVA film at 120○C for 2 h in a hot air oven. For MW crosslinking, a total of seven doses (each of 2 min duration) of 500 W MW power (2.45 GHz MW frequency) by LG solo MW model no. MS2021CW were given.
Preparation of BCNW–PVA and BCNW–TA–PVA Biocompo- site. Total volume of 25 mL BCNW/water suspension (5 mg mL−1) was added in 75 mL water and dispersed for 1 h using ultrasonic cleaning system (model no. EN-50-US; Lifecare Equip- ments Pvt. Ltd., Mumbai, India) followed by 30 min stirring. The BCNW/water mixture was heated at 90 ℃ for 1 h, and then PVA was dissolved into it. For BCNW–TA–PVA biocom- posite, 875 mg (35% w/w) TA was mixed after complete dissolu- tion of PVA. After drying, the BCNW–TA–PVA biocomposite was crosslinked similar to TA–PVA films by both MW irradia- tion and CH.
3.2 Characterization of Films
For Fourier transform infrared spectroscopy (FTIR), 20 mg sam- ples were ground with 80 mg of KBr and compressed into pellets. The FTIR spectra of the different PVA samples were recorded over the frequency range 400–4000 cm−1 using PerkinElmer spectrum version 10.03.06 instrument. Swelling study was per- formed to measure the amount of absorbed water according to the ISO standard (ISO 62:2008, Plastics—Determination of water absorption). Samples were cut into three pieces each of dimen- sion (2.5 cm × 2.5 cm), weighed, and immersed in DI water for 48 h at 25 . The swollen samples were taken out and wiped with tissue paper to remove the excess water and weighed. Tensile testing of the PVA films was performed using Instron universal testing machine model no. 3345 with a load cell of 5 kN. ASTM D 882-12 standard was used for testing of the films. All samples were cut into 10 rectangular strips of dimensions (70 mm × 10 mm) with 40 mm as gauge length and stretched at 10 mm min−1 at 50% relative humidity (RH) following the above standard. Samples were kept at 45℃ in a hot air oven to remove moisture before tensile testing. Additionally, to observe the effect of RH, samples were conditioned at 50% RH for 2 days. Statistical study was performed to observe the significance of the crosslinking method (CH and MW) on swelling and mechanical properties of crosslinked and biocomposite samples. The Tukey test in conju- gation with one-way analysis of variance (ANOVA) was per- formed using Microsoft Excel software. On the basis of p-value measured at 5% significant level, the tensile and swelling data were analyzed as reported in earlier studies.32 Thermogravimetric analysis (TGA) of PVA samples was performed by using TA Instruments model no. SDT Q600 V20.9. The TGA study was performed in the range of 30–600 ℃ with a heating rate of 10 ℃ min−1 under nitrogen gas. Field emission scanning electron microscopy (SEM) was performed by using the TESCAN MIRA3 instrument at 15 kV in SE mode to examine the BCNW and cell proliferation on biocomposite samples. The samples were gold coated before microscopy. To ascertain wettability of PVA sam- ples, water contact angle measurements were performed using Dataphysics OCA 35 goniometer equipped with a camera and software analyzer (Germany). The measurements were performed by putting one drop (10 μL) of water onto the surface with a syringe. Three readings of each sample at different positions were measured, and the average values of the contact angle were calcu- lated. For hemocompatibility assay, 50 mL blood of a freshly slaughtered goat was taken and mixed with 0.5 mL of 0.5 mM EDTA as anticoagulant. Chelated blood suspension (3 mL) was centrifuged at 700g for 10 min at room temperature, and the obtained RBC pellet was suspended in cold phosphate-buffered saline (PBS, pH 7.4). After three washes in cold PBS at 700g for 10 min, the obtained pellet of RBC was resuspended in 5 mL of cold PBS solution. This RBC suspension was always freshly pre- pared and used within 24 h of preparation. For hemolysis, 20 mg of prewashed and PBS equilibrated samples were transferred to 2 mL vials containing 200 μL cold PBS. Total volume of 800 μL RBC suspension was added to the samples previously incubated in PBS and incubated at 37 ℃ for 90 min in incubator shaker. After incubation, tubes were centrifuged at 700g for 10 min, and the absorbance of the supernatant was analyzed using ultraviolet (UV) spectrophotometer at 540 nm for the release of hemoglobin in the solution. For positive control, 0.2% Triton-X 100 was added to the RBC suspension leading to complete hemolysis and supernatant absorbance was measured after centrifuging at 700g for 10 min. The experiment was repeated twice (n = 3) with washed and unwashed samples, and the average value was taken. In vitro cell culture: L929 mouse fibroblast cells were cultured in high glucose DMEM supplemented with 10% fetal bovine serum and 1% antibiotic solution (penicillin and streptomycin) (com- plete medium). The cells were cultured in two-dimensional (2D) tissue culture plates (TCPs) under a humidified environ- ment in an incubator at 37℃ with 5% CO2. The films were seeded with fibroblast cells to study in vitro cell–material interac- tion. The composite samples prepared using CH and MW were cut into 1 cm × 1 cm pieces and washed overnight (on a mechanical shaker) with methanol to remove the unreacted TA. The samples were then sterilized by placing them in 70% ethanol for 24 h at room temperature. Sterilized films were equili- brated in 1 × PBS (pH 7.4) and 1% antibiotic (penicillin and streptomycin) for 12 h. The cells were harvested from the 2D TCP using trypsin–EDTA solution. The viability of the cells was checked using a trypan blue exclusion assay. Then, 1 × 104 cells were dispersed in 20 μL of complete DMEM and seeded onto the PVA samples. An equal number of cells were seeded onto 24-well 2D TCP as a positive control. The media in plates and wells were replenished every 48 h. Cell proliferation MTT assay using L929 mouse fibroblast cells: The cell culture on biocomposite samples prepared by CH and MW crosslinking was performed for 16 days to check cell proliferation and viability. The readings of MTT assays among the groups were normalized for surface area. For MTT assay, the samples were transferred into fresh wells and washed with 1 × PBS. The samples were then incubated for 4 h in 500 μL of incomplete DMEM medium containing MTT reagent (0.5 mg/mL). The color change was measured at 570 nm using UV–visible spectrophotometer (Evolution 201; Thermo Sci- entific) using dimethyl sulfoxide as blank. Cell–material interaction by SEM imaging: To check the cell–material interac- tion (adhesion and proliferation), the samples were harvested 24 h after seeding fibroblast cells on them, washed thrice with 1 × PBS, and fixed using glutaraldehyde (2 % v/v) solution at 4 ℃ for 4 h. The samples were then freeze-dried to remove mois- ture and gold coated for SEM analysis.
4 RESULTS AND DISCUSSION
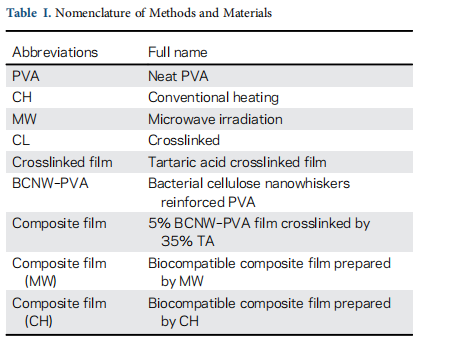
In this article, the properties of composite films prepared by crosslinking 5% BCNW–PVA by 35% w/w TA using CH and MW irradiation are compared. Additionally, hemocompatibility and cytocompatibility of the composites are also studied. For convenience, the nomenclature of method and materials is men- tioned in Table I.
4.1 FTIR Spectroscopy—Identification of Crosslink Bonds
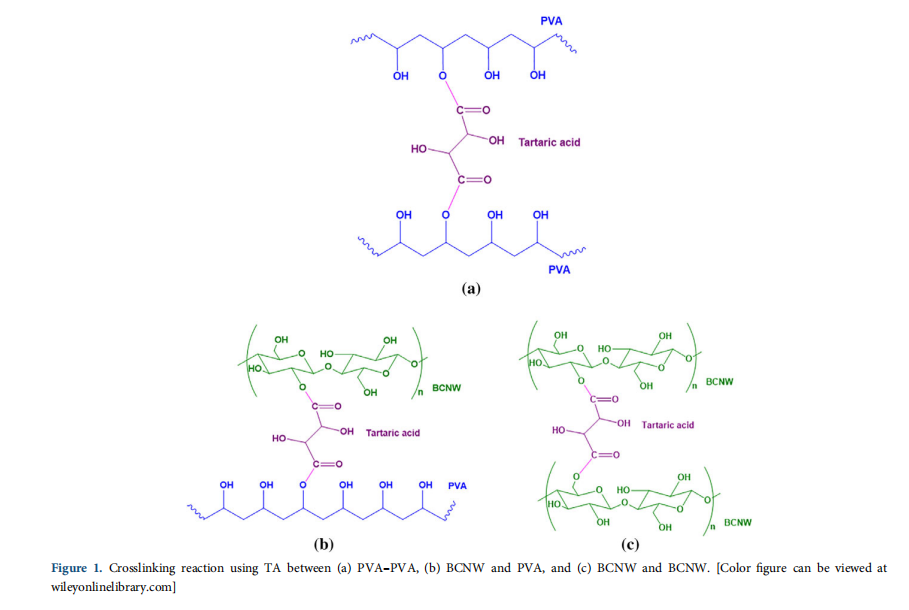
The crosslinking reaction used here is solid-state esterification where hydroxyl groups of PVA and cellulose react with carboxylic groups of TA to form tartrate crosslink networks in PVA. Similar to BCNW–PVA [Figure 1(b)], crosslinking can also occur between PVA–PVA [Figure 1(a)] and BCNW–BCNW [Figure 1(c)].
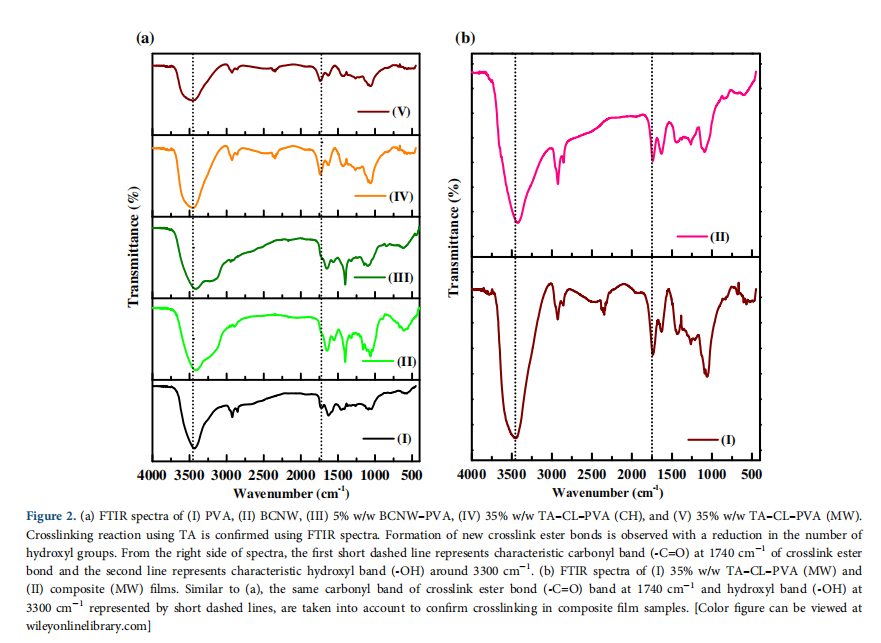
PVA has characteristics absorption bands at 3200–3400 cm−1 (–OH stretching), 2940 cm−1 (–CH stretching), 1640 cm−1 (absorbed water),36 1435 cm−1 (–CH bending), and 1095 cm−1 (–C–O stretching)37 as shown in Figure 2. As PVA is obtained from the hydrolysis of poly(vinyl acetate), it has characteristic small absorption carbonyl band (–C=O) peak due to unhydro- lyzed acetate groups. Crosslinking results in the formation of tar- trate crosslinks at the expense of hydroxyl groups leading to an increase of carbonyl bond intensity at the expense of hydroxyl band. Hence, the ratio of (–C=O)/(–OH) bands for crosslinked and noncrosslinked samples is measured to confirm crosslinking as described earlier.27,28,30,31 For 35% w/w TA crosslinked PVA prepared by CH and MW methods, the values of intensity ratio (–C=O/–OH) are measured as 0.5 and 0.53 respectively, which is higher compared to PVA (0.34), confirming crosslinking. The FTIR spectra of the composite film in Figure 2(b) are similar to crosslinked PVA confirming crosslinking.
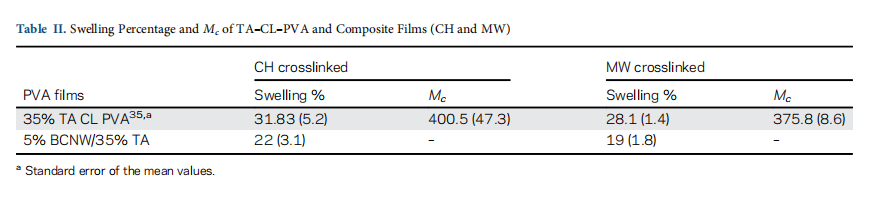
4.2 Swelling Study (Water Uptake)
The water absorption of composite films prepared by CH a MW method is measured as swelling percentage using t equation:
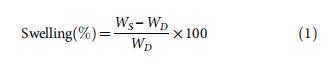
where WS is the weight of swollen films, and WD is the weight dry films.
The value of water absorption in terms of swelling percentage crosslinked PVA and composite samples (CH and MW) are mtioned in Table II. PVA has a higher value of swelling percenta of 203% due to the presence of a large number of hydrox groups. These hydroxyl groups allow incoming water to occu the interspaces between mobile polymer chains in the amorpho region of polymer and hence cause large swelling. In crosslink films, less number of hydroxyl groups and restricted chain mo ment results in significantly lower water absorption compar to PVA.
In composite films, crosslinking of BCNW–PVA by TA also deonstrate significant lowering in the water absorption of PVA. T composite films prepared by MW has the least water uptake 19% (p-value <0.05), which is slightly better than the swelling pcentage of composite films 23% (p-value <0.05) prepared by CH.
4.3 Tensile Testing
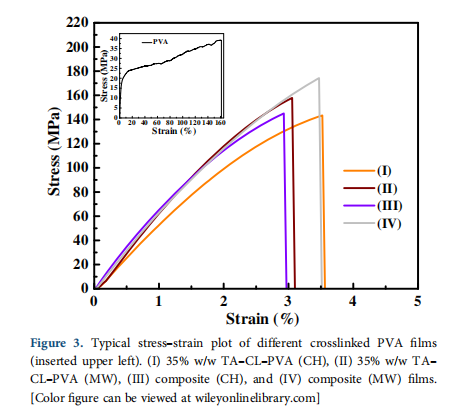
The stress–strain plot of crosslinked (CH and MW) and compos- ite (CH and MW) samples follow a similar trend and is different from that of PVA (Figure 3). The mechanical properties, namely, tensile strength and elongation at break of PVA, crosslinked PVA and composite films at 0 and 50% RH are summarized in Table III.
Crosslinking improves the interfacial interaction between rein- forcement BCNW and PVA matrix from hydrogen to covalent bonding [Figure 1(b)] thereby providing synergy between cross- linking and reinforcement resulting in the higher tensile strength of composite films. At 0% RH, composite films (CH) demonstrate the strength of 159.1 MPa compared to 5% BCNW–PVA (85 MPa) and PVA (42 MPa) at the expense of higher elongation at break for PVA (120%). Composite films (MW) show higher strength at 0% RH (172 MPa, p-value <0.05), which is even better than that of composite films (CH). It could be due to homoge- neous heating of the polymer by MW during the crosslinking reac- tion. The acquired value of biocomposite is highest among the published reports based on similar materials and methods.
Crosslinked PVA samples primed at 50% RH exhibit reduced strength compared to those at 0% RH, which could be due to the plasticizing role of the absorbed water. However, it must be noted that composite films (MW and CH) demonstrate better retention of
strength compared to crosslinked (35% TA–CL–PVA) samples. The retention in the strength of CH and MW biocomposite 89 and 93% respectively, is much higher compared to 35% TA–CL–PVA, which is 45% for CH and 40% for MW samples.35 This could be due to the plasticizing role of absorbed moisture by uncrosslinked acid at 50% RH.35,39 The absorbed moisture reduces the strength and increases the elongation at break values of crosslinked samples. In the CH biocomposite, crosslinking is possible between the PVA–PVA, BCNW–BCNW, and BCNW–PVA. Hence, most of the crosslinker is consumed in crosslinking and less plasticizing effect due to unreacted acid is observed in comparison to 35% TA–CL–PVA (CH). On comparing the difference in strength of composite films (CH and MW) measured at different RHs (Table III), MW biocomposite shows better stability in the strength due to effec- tive crosslinking.
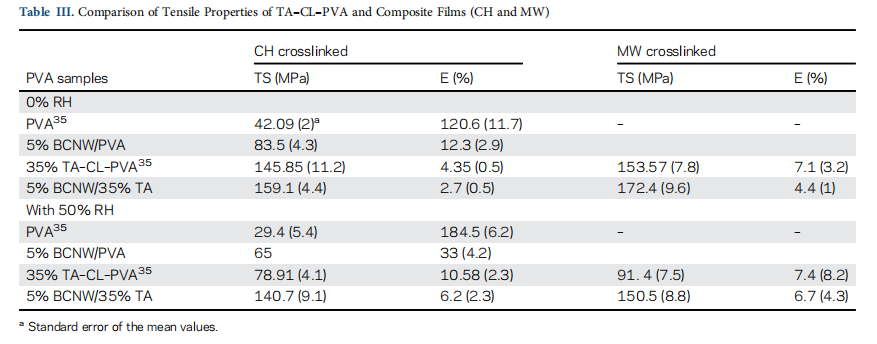
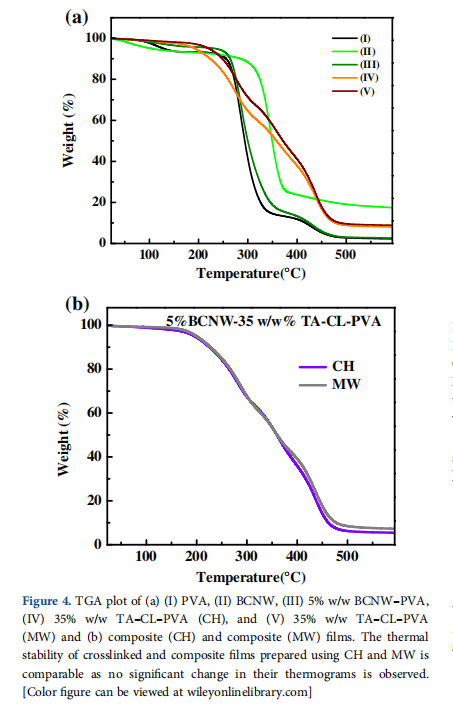
4.4TGA—Thermal Stability
The thermal decomposition of PVA is observed over three tem- perature ranges.31 In all the three stages of thermal degradation of PVA [Figure 4(a)]; 80–250 ℃ corresponded to removal of bound and unbound water molecules of PVA, 275–450 ℃ to chain scission and degradation of the PVA backbone and 475– 525 ○C to the left over weight of charred residues of black carbon.
In the first degradation stage, the composite samples (CH and MW) have less absorbed water and hydroxyl groups due to chemical crosslinking between PVA and cellulose. Therefore,
omposite samples show less weight loss in comparison to PVA and BCNW–PVA. Furthermore, crosslinking modifies the PVA backbone with the formation of tartrate crosslinks. As a result crosslinked samples have 40–50% weight remaining compared to BCNW–PVA and PVA (10–12% left over weight) in the second stage. Similarly, in the third degradation stage, PVA shows 95–97% weight loss, but crosslinked samples indicate weight loss o 85–87%.
Figure 4(a) shows that BCNW possesses higher thermal stability in comparison to other samples. It is due to the strongly inter connected network of H-bonding between cellulose fibrils. Therefore, it is expected that BCNW could contribute to improving the thermal stability of reinforced PVA samples. Nevertheless, PVA and BCNW–PVA samples have shown similar thermal stability. It can be explained as they have an only H-bonding interfacial binding that is incapable of showing thermal resistance at higher temperatures like in the case of crosslinked PVA samples.28,31.In composite (CH and MW) samples, crosslinking results in the reduction of hydroxyl groups with the formation of new covalent crosslinks in the BCNW–PVA structure. Therefore, crosslinking shifts the degradation temperature of BCNW–PVA samples to higher temperatures [Figure 4(a,b)]. However, the thermal degradation behavior of biocomposite samples is found to be similar to the crosslinked PVA samples [Figure 4(b)]. In addition, no difference is observed in their thermal stability of biocomposites prepared by CH or MW.
4.5 Hemocompatibility
The hemocompatibility of bacterial cellulose and PVA is well established.5 However, it is undeniable that new chemical bonds formed by crosslinking PVA and BCNW–PVA can change the hemocompatibility of the parent materials (PVA and bacterial cel-lulose). Hence, hemocompatibility of crosslinked and biocomposite
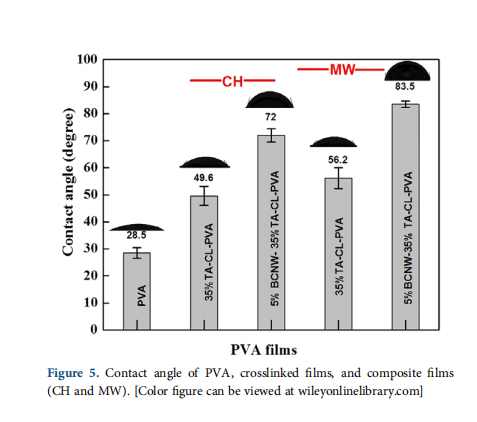
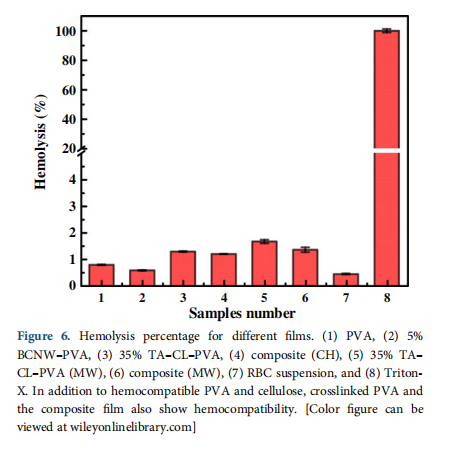
samples prepared by CH and MW is measured (Figure 6). Hemolysis percentage is calculated by eq. (2):

where OD is the optical density of the samples, measured at 540 nm. The crosslinked PVA and biocomposite samples show negative hemolysis (<5%) results and therefore, are hemocompatible.
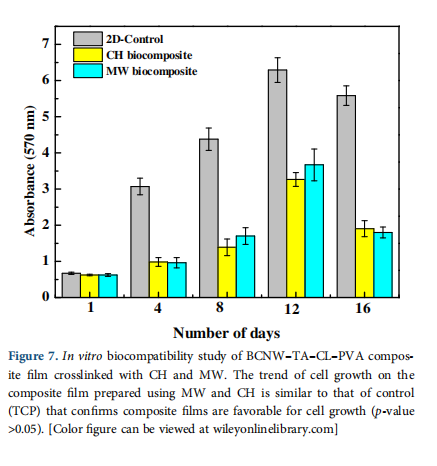

4.6 Biocompatibility—In Vitro Test
The MTT results for Day 1 (Figure 7) show uniform seeding on the films with no differences among groups. An increase in the MTT reading on Day 8 is observed showing positive cell growth.
The MTT values show a positive increase up to Day 12 with a fourfold increase in MTT values on composite films compared to Day 1 values. This confirms that the biocomposite samples support cell adhesion, proliferation and are biocompatible. Subsequently, at Day 16, a decrease in MTT reading is observed. This could be due to cell detachment at later time points due to cell over confluence on the composite film surfaces. The 2D control TCP shows the highest MTT readings at all-time points.
4.7 SEM Imaging
SEM imaging is used to examine the BCNW formation and cell growth over biocomposite surfaces prepared by MW and CH crosslinking. Figure 8(a) is an SEM image of several microns length cellulose fibrils with 70–100 nm diameter arranged in random order. The conversion of bacterial cellulose results in the shortening of cellulose fibrils shown by Figure 8(b). Thebiocomposite samples prepared by CH and MW are biocompatible and favorable for cell growth. In Figure 8(c,d), SEM images show fibroblast growth over the films confirming that cells are properly dispersed over the surface of the MW biocomposite. Filopodial focal adhesion complexes that are necessary for their adhesion and migration are also observed from the cells on films. SEM imaging shows that the MW biocomposite shows positive cell–material interaction, supports cell growth, that is also confirmed by hemocompatibility and MTT assay.
5 REFERENCES
- Qiu, K.; Netravali, A. N. Compos. Sci. Technol. 2012, 72, 1588.
- Qiu, K.; Netravali, A. N. J. Mater. Sci. 2012, 47, 6066.
- Zheng, Q.; Cai, Z.; Gong, S. J. Mater. Chem. A. 2014, 2, 3110.Wang, L.-Y.;
- Wang, M.-J. ACS Sustain. Chem. Eng. 2016, 4, 2830.
- Leitão, A. F.; Gupta, S.; Silva, J. P.; Reviakine, I.; Gama, M. Colloids Surf. B Biointerfaces. 2013, 111, 493.
- Millon, L. E.; Guhados, G.; Wan, W. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2008, 86B, 444.
- Wang, J.; Gao, C.; Zhang, Y.; Wan, Y. Mater. Sci. Eng. C. 2010, 30, 214.
- Tummala, G. K.; Rojas, R.; Mihranyan, A. J. Phys. Chem. B. 2016, 120, 13094.
- Tummala, G. K.; Joffre, T.; Lopes, V. R.; Liszka, A.; Buznyk, O.; Ferraz, N.; Persson, C.; Griffith, M.; Mihranyan, A. ACS Biomater. Sci. Eng. 2016, 2, 2072.
- Hallensleben, M. L. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: New York, 2000.
- Mohanty, S.; Larsen, L. B.; Trifol, J.; Szabo, P.; Burri, H. V. R.; Canali, C.; Dufva, M.; Emnéus, J.; Wolff, A. Mater. Sci. Eng. C. 2015, 55, 569.
- Jiang, S.; Liu, S.; Feng, W. J. Mech. Behav. Biomed. Mater. 2011, 4, 1228.
- Stammen, J. A.; Williams, S.; Ku, D. N.; Guldberg, R. E. Biomaterials. 2001, 22, 799.
- Chu, K. C.; Rutt, B. K. Magn. Reson. Med. 1997, 37, 314.
- Doblin, M. S.; Kurek, I.; Jacob-Wilk, D.; Delmer, D. P.
- Plant Cell Physiol. 2002, 43, 1407.
- Guo, J.; Catchmark, J. M. Carbohydr. Polym. 2012, 87, 1026.
- Sakairi, N.; Asano, H.; Ogawa, M.; Nishi, N.; Tokura, S. Carbohydr. Polym. 1998, 35, 233.
- Svensson, A.; Nicklasson, E.; Harrah, T.; Panilaitis, B.; Kaplan, D. L.; Brittberg, M.; Gatenholm, P. Biomaterials. 2005, 26, 419.
- Bäckdahl, H.; Helenius, G.; Bodin, A.; Nannmark, U.; Johansson, B. R.; Risberg, B.; Gatenholm, P. Biomaterials. 2006, 27, 2141.
- Lv, X.; Yang, J.; Feng, C.; Li, Z.; Chen, S.; Xie, M.; Huang,J.; Li, H.; Wang, H.; Xu, Y. ACS Biomater. Sci.Eng. 2016, 2, 19.
- Awadhiya, A.; Kumar, D.; Rathore, K.; Fatma, B.; Verma,V. Polym. Bull. 2017, 74, 2887.
- Millon, L. E.; Oates, C. J.; Wan, W. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2009, 90B, 922.
- Song, T.; Tanpichai, S.; Oksman, K. Cellulose. 2016, 23, 1925.
- Virtanen, S.; Vartianen, J.; Setala, H.; Tammelin, T.; Vuoti,S. RSC Adv. 2014, 4, 11343.
- Lee, S.-Y.; Mohan, D. J.; Kang, I.-A.; Doh, G.-H.; Lee, S.; Han,S. O. Fibers Polym. 2009, 10, 77.
- Martínez-Sanz, M.; Lopez-Rubio, A.; Lagaron, J. M. Carbohydr. Polym. 2011, 85, 228.
- Sonker, A. K.; Rathore, K.; Nagarale, R. K.; Verma, V. J. Polym. Environ. 2018, 26, 1782.
- Sonker, A. K.; Tiwari, N.; Nagarale, R. K.; Verma, V. J. Polym. Sci. Part A: Polym. Chem. 2016, 54, 2515.
- Sonker, A. K.; Wagner, H. D.; Bajpai, R.; Tenne, R.; Sui, X. Compos. Sci. Technol. 2016, 127, 47.
- Gohil, J. M.; Bhattacharya, A.; Ray, P. J. Polym. Res. 2006, 13, 161.
- Sonker, A. K.; Teotia, A. K.; Kumar, A.; Nagarale, R. K.; Verma, V. Macromol. Chem. Phys. 2017, 218, 1700130.
- Awadhiya, A.; Kumar, D.; Verma, V. Carbohydr. Polym. 2016, 151, 60.
- Krumova, M.; López, D.; Benavente, R.; Mijangos, C.; Pereña, J. M. Polymer. 2000, 41, 9265.
- 34. Xia, L. L.; Li, C. L.; Wang, Y. J. Membr. Sci. 2016, 498, 263.
- Sonker, A. K.; Verma, V. J. Appl. Polym. Sci. 2018, 135, 46125.
- Kim, S.-K.; Wie, J. J.; Mahmood, Q.; Park, H. S. Nanoscale. 2014, 6, 7430.
- Blout, E. R.; Karplus, R. J. Am. Chem. Soc. 1948, 70, 862.
- Niu, Y.; Zhang, X.; He, X.; Zhao, J.; Zhang, W.; Lu, C. Int. J. Biol. Macromol. 2015, 72, 855.
- Belay, M.; Sonker, A. K.; Nagarale, R. K.; Verma, V. Polym. Int. 2017, 66, 1737.
- van Wachem, P. B.; Beugeling, T.; Feijen, J.; Bantjes, A.; Detmers, J. P.; van Aken, W. G. Biomaterials. 1985, 6, 403.
- van Kooten, T. G.; Schakenraad, J. M.; van der Mei, H. C.; Busscher, H. J. Biomaterials. 1992, 13, 897.
- Thomas, L. V.; Arun, U.; Remya, S.; Nair, P. D. J. Mater. Sci. Mater. Med. 2008, 20, 259.

